Disulfide bond
In chemistry, a disulfide (Br.E. disulphide) bond is a covalent bond, usually derived by the coupling of two thiol groups. The linkage is also called an SS-bond or disulfide bridge. The overall connectivity is therefore R-S-S-R. The terminology is widely used in biochemistry. Formally the connection is called a persulfide, in analogy to its congener, a peroxide (R-O-O-R), but this terminology is obscure.
Contents |
Properties
The disulfide bond is strong, a typical bond dissociation energy being 60 kcal/mole. Being about 40% weaker than C-C and C-H bonds, the disulfide bond is thus often the "weak link" in many molecules. Furthermore, reflecting the polarizability of divalent sulfur, the S-S bond is susceptible to scission by polar reagents, both electrophiles and especially nucleophiles:[1]
- RS-SR + Nu- → RS-Nu + RS-
The disulfide bond is about 2.05 Å in length, about 0.5 Å longer than a C-C bond. Rotation about the S-S axis is subject to a low barrier. Disulfides show a distinct preference for dihedral angles approaching 90°. When the angle approaches 0° or 180°, then the disulfide is a significantly better oxidant.
Disulfides where the two R groups are the same are called symmetric, examples being diphenyl disulfide and dimethyl disulfide. When the two R groups are not identical, the compound is said to be an unsymmetric or mixed disulfide.[2]
Although the hydrogenation of disulfides is usually not practical, the equilibrium constant for the reaction provides a measure of the standard redox potential for disulfides:
- RSSR + H2 → 2 RSH
This value is about -250 mV vs NHE (pH = 7). By comparison, the standard reduction potential for ferrodoxins is about -430 mV.
Preparation and reactions
Formation of disulfides
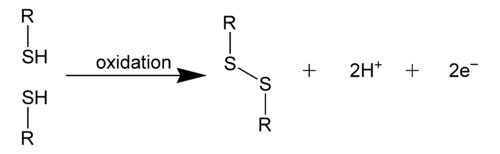
Disulfide bonds are usually formed from the oxidation of sulfhydryl (-SH) groups, especially in biological contexts.[3] The transformation is depicted as follows:
- 2 RSH → RS-SR + 2 H+ + 2 e-
A variety of oxidants promote this reaction including air and hydrogen peroxide. Such reactions are thought to proceed via sulfenic acid intermediates. In the laboratory, iodine in the presence of base is commonly employed to oxidize thiols to disulfides. Several metals, such as copper(II) and iron(III) complexes effect this reaction. Alternatively, disulfide bonds in proteins often formed by thiol-disulfide exchange:
- RS-SR + R'SH
 R'S-SR + RSH
R'S-SR + RSH
Such reactions are mediated by enzymes in some cases and in other cases are under equilibrium control, especially in the presence of catalytic amount of base.
The alkylation of alkali metal di- and polysulfides gives disulfides. "Thiokol" polymers arise when sodium polysulfide is treated with an alkyl dihalide. In the converse reaction, carbanionic reagents react with elemental sulfur to afford mixtures of the thioether, disulfide, and higher polysulfides. These reactions are often unselective but can been optimized for specific applications.
Many specialized methods have been developed for forming disulfides, usually for applications in organic synthesis. Reagents that deliver the equivalent of "RS+" react with thiols to give unsymmetrical disulfides:[3]
- RSH + R'SNR"2 → RS-SR' + HNR"2, where R"2N = phthalimido
Scission of disulfides
The most important reaction of disulfide bonds is their cleavage, which occurs via reduction. A variety of reductants can be used. In biochemistry, thiols such as mercaptoethanol or dithiothreitol serve as reductants, the thiol reagents are used in excess to drive the equilibrium to the right:
- RS-SR + HOCH2CH2SH HOCH2CH2S-SCH2CH2OH + 2 RSH
In organic synthesis, hydride agents are typically employed for scission of disulfides, such as sodium borohydride. More aggressive, alkali metals will affect this reaction:
- RS-SR + 2 Na → 2 NaSR
These reactions are often followed by protonation of the resulting metal thiolate:
- NaSR + HCl → HSR + NaCl
Thiol-disulfide exchange is a chemical reaction in which a thiolate group 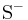 attacks a sulfur atom of a disulfide bond -S-S-. The original disulfide bond is broken, and its other sulfur atom (green atom in Figure 1) is released as a new thiolate, carrying away the negative charge. Meanwhile, a new disulfide bond forms between the attacking thiolate (red atom in Figure 1) and the original sulfur atom (blue atom in Figure 1).[4][5]
attacks a sulfur atom of a disulfide bond -S-S-. The original disulfide bond is broken, and its other sulfur atom (green atom in Figure 1) is released as a new thiolate, carrying away the negative charge. Meanwhile, a new disulfide bond forms between the attacking thiolate (red atom in Figure 1) and the original sulfur atom (blue atom in Figure 1).[4][5]
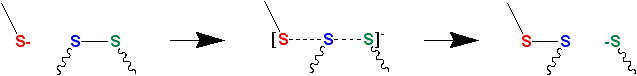
Thiolates, not thiols, attack disulfide bonds. Hence, thiol-disulfide exchange is inhibited at low pH (typically, below 8) where the protonated thiol form is favored relative to the deprotonated thiolate form. (The pKa of a typical thiol group is roughly 8.3, but can vary due to its environment.)
Thiol-disulfide exchange is the principal reaction by which disulfide bonds are formed and rearranged in a protein. The rearrangement of disulfide bonds within a protein generally occurs via intra-protein thiol-disulfide exchange reactions; a thiolate group of a cysteine residue attacks one of the protein's own disulfide bonds. This process of disulfide rearrangement (known as disulfide shuffling) does not change the number of disulfide bonds within a protein, merely their location (i.e., which cysteines are bonded). Disulfide reshuffling is generally much faster than oxidation/reduction reactions, which change the number of disulfide bonds within a protein. The oxidation and reduction of protein disulfide bonds in vitro also generally occurs via thiol-disulfide exchange reactions. Typically, the thiolate of a redox reagent such as glutathione or dithiothreitol attacks the disulfide bond on a protein forming a mixed disulfide bond between the protein and the reagent. This mixed disulfide bond when attacked by another thiolate from the reagent, leaves the cysteine oxidised. In effect, the disulfide bond is transferred from the protein to the reagent in two steps, both thiol-disulfide exchange reactions.
The in vivo oxidation and reduction of protein disulfide bonds by thiol-disulfide exchange is facilitated by a protein called thioredoxin.
Other reactions of disulfides
Many specialized organic reactions have been developed for disulfides, again mainly associated with the scission of the S-S bond, which is usually the weakest bond in a molecule. In the Zincke disulfide cleavage reactions, disulfides are cleaved to give the to a sulfenyl halide by reaction with bromine or chlorine.[6][7]
Occurrence in proteins
Disulfide bonds play an important role in the folding and stability of some proteins, usually proteins secreted to the extracellular medium.[2] Since most cellular compartments are reducing environments, disulfide bonds are generally unstable in the cytosol with some exceptions as noted below.
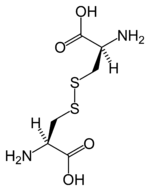
Disulfide bonds in proteins are formed between the thiol groups of cysteine residues. The other sulfur-containing amino acid, methionine, cannot form disulfide bonds. A disulfide bond is typically denoted by hyphenating the abbreviations for cysteine, e.g., when referring to Ribonuclease A the "Cys26-Cys84 disulfide bond", or the "26-84 disulfide bond", or most simply as "C26-C84"[8] where the disulfide bond is understood and does not need to be mentioned. The prototype of a protein disulfide bond is the two-amino-acid peptide, cystine, which is composed of two cysteine amino acids joined by a disulfide bond (shown in Figure 2 in its unionized form). The structure of a disulfide bond can be described by its 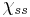 dihedral angle between the
dihedral angle between the 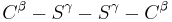 atoms, which is usually close to ±90°.
atoms, which is usually close to ±90°.
The disulfide bond stabilizes the folded form of a protein in several ways:
1) It holds two portions of the protein together, biasing the protein towards the folded topology. Stated differently, the disulfide bond destabilizes the unfolded form of the protein by lowering its entropy.
2) The disulfide bond may form the nucleus of a hydrophobic core of the folded protein, i.e., local hydrophobic residues may condense around the disulfide bond and onto each other through hydrophobic interactions.
3) Related to #1 and #2, the disulfide bond link two segments of the protein chain, the disulfide bond increases the effective local concentration of protein residues and lowers the effective local concentration of water molecules. Since water molecules attack amide-amide hydrogen bonds and break up secondary structure, a disulfide bond stabilizes secondary structure in its vicinity. For example, researchers have identified several pairs of peptides that are unstructured in isolation, but adopt stable secondary and tertiary structure upon forming a disulfide bond between them.
A disulfide species is a particular pairing of cysteines in a disulfide-bonded protein and is usually depicted by listing the disulfide bonds in parentheses, e.g., the "(26-84, 58-110) disulfide species". A disulfide ensemble is a grouping of all disulfide species with the same number of disulfide bonds, and is usually denoted as the 1S ensemble, the 2S ensemble, etc. for disulfide species having one, two, etc. disulfide bonds. Thus, the (26-84) disulfide species belongs to the 1S ensemble, whereas the (26-84, 58-110) species belongs to the 2S ensemble. The single species with no disulfide bonds is usually denoted as R for "fully reduced". Under typical conditions, disulfide reshuffling is much faster than the formation of new disulfide bonds or their reduction; hence, the disulfide species within an ensemble equilibrate more quickly than between ensembles.
The native form of a protein is usually a single disulfide species, although some proteins may cycle between a few disulfide states as part of their function, e.g., thioredoxin. In proteins with more than two cysteines, non-native disulfide species may be formed, which are almost always unfolded. As the number of cysteines increases, the number of nonnative species increases factorially. The number of ways of forming p disulfide bonds from n cysteine residues is given by the formula
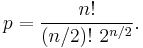
For example, an eight-cysteine protein such as ribonuclease A has 105 different four-disulfide species, only one of which is the native disulfide species. Isomerases have been identified that catalyze the interconversion of disulfide species, accelerating the formation of the native disulfide species.
Disulfide species that have only native disulfide bonds (but not all of them) are denoted by des followed by the lacking native disulfide bond(s) in square brackets. For example, the des[40-95] disulfide species has all the native disulfide bonds except that between cysteines 40 and 95. Disulfide species that lack one native disulfide bond are frequently folded, particularly if the missing disulfide bond is exposed to solvent in the folded, native protein.
In order to analyze the structure of proteins, it is often necessary to break disulfide bonds. This reduction of disulfide bonds can be accomplished by treatment with 2-mercaptoethanol, dithiothreitol, or tris(2-carboxyethyl)phosphine.
In prokaryotes and archaea
Disulfide bonds play an important protective role for bacteria as a reversible switch that turns a protein on or off when bacterial cells are exposed to oxidation reactions. Hydrogen peroxide (H2O2) in particular could severely damage DNA and kill the bacterium at low concentrations if not for the protective action of the SS-bond. Archaea typically have fewer disulfides than higher organisms.[9]
In eukaryotes
In eukaryotic cells, disulfide bonds are generally formed in the lumen of the RER (rough endoplasmic reticulum) but not in the cytosol. This is due to the oxidative environment of the ER and the reducing environment of the cytosol (see glutathione). Thus disulfide bonds are mostly found in secretory proteins, lysosomal proteins, and the exoplasmic domains of membrane proteins.
Notable exceptions to this rule include a number of cytosolic proteins have cysteine residues in proximity to each other that function as oxidation sensors; when the reductive potential of the cell fails, they oxidize and trigger cellular response mechanisms. Vaccinia virus also produces cytosolic proteins and peptides that have many disulfide bonds; although the reason for this is unknown presumably they have protective effects against intracellular proteolysis machinery.
Disulfide bonds are also formed within and between protamines in the sperm chromatin of many mammalian species.
Disulfides in regulatory proteins
As disulfide bonds can be reversibly reduced and re-oxidized, the redox state of these bonds has evolved into a signaling element. In chloroplasts, for example, the enzymatic reduction of disulfide bonds has been linked to the control of numerous metabolic pathways as well as gene expression. The reductive signaling activity has been shown, thus far, to be carried by the ferredoxin thioredoxin system, channeling electrons from the light reactions of photosystem I to catalytically reduce disulfides in regulated proteins in a light dependent manner. In this way chloroplasts adjust the activity of key processes such as the Calvin-Benson cycle, starch degradation, ATP production and gene expression according to light intensity.
In hair and feathers
Over 90% of the dry weight of hair are proteins called keratins, which have a high disulfide content, from the amino acid cysteine. The robustness conferred in part by disulfide linkages is illustrated by the recovery of virtually intact hair from ancient Egyptian tombs. Feathers have similar keratins and are extremely resistant to protein digestive enzymes. Different parts of the hair and feather have different cysteine levels, leading to harder or softer material. Manipulating disulfide bonds in hair is the basis for the permanent wave in hairstyling. Reagents that affect the making and breaking of S-S bonds are key, e.g. ammonium thioglycolate. The high disulfide content of feathers dictates the high sulfur content of bird eggs. The high disulfide content of hair and feathers contributes to the disagreeable odor that results when they are burned.
In industry
Disulfide and (polysulfide) bonds are the crosslinking groups that result from the vulcanization of rubber. In analogy to the role of disulfides in proteins, the S-S linkages in rubber are crosslinkers, and strongly affect the rheology of the material.
Related compounds
Thiuram disulfides, with the formula (R2NC(S)S)2, are disulfides but they behave distinctly because of the thiocarbonyl group. Disulfides are analogous but more common that the related compounds peroxides and diselenides. Compounds with three sulfur atoms, e.g. CH3S-S-SCH3, are called trisulfides. Isomeric with disulfides are thiosulfoxides.
References
- ↑ R. J. Cremlyn “An Introduction to Organosulfur Chemistry” John Wiley and Sons: Chichester (1996). ISBN 0 471 95512 4.
- ↑ 2.0 2.1 Sevier, C. S. and Kaiser, C. A. (2002). "Formation and transfer of disulphide bonds in living cells". Nature Reviews Molecular and Cellular Biology 3: 836–847.
- ↑ 3.0 3.1 Witt, D. (2008). "Recent developments in disulfide bond formation". Synthesis: 2491–2509. doi:10.1055/s-2008-1067188.
- ↑ Gilbert HF (1990). "Molecular and Cellular Aspects of Thiol-Disulfide Exchange". Advances in Enzymology 63: 69–172. doi:10.1002/9780470123096.ch2. PMID 2407068.
- ↑ Gilbert HF (1995). "Thiol/disulfide exchange equilibria and disulfide bond stability". Methods in Enzymology 251: 8–28. doi:10.1016/0076-6879(95)51107-5.
- ↑ For example the conversion of di-o-nitrophenyl disulfide to o-nitrophenylsulfur chloride Max H. Hubacher (1943), "o-Nitrophenylsulfur chloride", Org. Synth., http://www.orgsyn.org/orgsyn/orgsyn/prepContent.asp?prep=CV2P0455; Coll. Vol. 2: 455
- ↑ Methyl sulfenyl chloride: Irwin B. Douglass and Richard V. Norton (1973), "Methanesulfinyl Chloride", Org. Synth., http://www.orgsyn.org/orgsyn/orgsyn/prepContent.asp?prep=CV5P0709; Coll. Vol. 5: 709
- ↑ Ruoppolo M, Vinci F, Klink TA, Raines RT, Marino G. (2000). "Contribution of individual disulfide bonds to the oxidative folding of ribonuclease A". Biochemistry 39 (39): 12033–42. PMID 11009618.
- ↑ Ladenstein, R. and Ren, B. (2008). "Reconsideration of an early dogma, saying "there is no evidence for disulfide bonds in proteins from archaea"". Extremophiles 12: 29–38. doi:10.1007/s00792-007-0076-z.
Further reading
- Sela M, Lifson S. (1959). "The reformation of disulfide bridges in proteins". Biochim Biophys Acta 36 (2): 471–8. doi:10.1016/0006-3002(59)90188-X. PMID 14444674.
- Stark GR.; Stern, K; Atala, A; Yoo, J (1977). "Cleavage at cysteine after cyanylation". Methods Enzymol 47: 129–32. doi:10.1016/j.ymeth.2008.09.005. PMID 927170.
- Thornton JM. (1981). "Disulphide bridges in globular proteins". J Mol Biol 151 (2): 261–87. doi:10.1016/0022-2836(81)90515-5. PMID 7338898.
- Thannhauser TW, Konishi Y, Scheraga HA. (1984). "Sensitive quantitative analysis of disulfide bonds in polypeptides and proteins". Anal Biochem 138 (1): 181–8. doi:10.1016/0003-2697(84)90786-3. PMID 6547275.
- Wu J, Watson JT. (1998). "Optimization of the cleavage reaction for cyanylated cysteinyl proteins for efficient and simplified mass mapping". Anal Biochem 258 (2): 268–76. doi:10.1006/abio.1998.2596. PMID 9570840.
- Futami J, Tada H, Seno M, Ishikami S, Yamada H. (2000). "Stabilization of human RNase 1 by introduction of a disulfide bond between residues 4 and 118". J Biochem 128 (2): 245–50. PMID 10920260. http://jb.oxfordjournals.org/cgi/content/abstract/128/2/245.
- Wittenberg, G, Danon, A (2008). "Disulfide bond formation in chloroplasts: Formation of disulfide bonds in signaling chloroplast proteins". Plant Science 175 (4): 459–466. doi:10.1016/j.plantsci.2008.05.011.
- Kadokura, Hiroshi; Katzen, Federico; Beckwith, Jon (2003). "Protein Disulfide Bond Formation in Prokaryotes". Annual Review of Biochemistry 72: 111. doi:10.1146/annurev.biochem.72.121801.161459.
- Tu, B. P. (2004). "Oxidative protein folding in eukaryotes: mechanisms and consequences". The Journal of Cell Biology 164: 341. doi:10.1083/jcb.200311055.
- Ellgaard, Lars; Ruddock, Lloyd W. (2005). "The human protein disulphide isomerase family: substrate interactions and functional properties". EMBO reports 6: 28. doi:10.1038/sj.embor.7400311.
External links
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||